ORA enrichment - single cell metabolomics
Bishoy Wadie
Source:vignettes/ORA_enrichment-single_cell_metabolomics.Rmd
ORA_enrichment-single_cell_metabolomics.RmdSingle-cell metabolomics generates output similar to scRNA-Seq, resulting in a matrix of metabolites (m) by cells (c), where values represent the abundance of each metabolite per cell. As with any single-cell analysis pipeline, enrichment analysis is typically one of the final steps to interpret the differential markers identified earlier.
Overrepresentation analysis (ORA) is the most common enrichment method for both bulk and single-cell datasets, regardless of the molecular readout. In metabolomics, several methods exist for metabolite enrichment, with MetaboAnalyst being the most popular. MetaboAnalyst is a robust platform offering various analyses for metabolomics data at different stages (raw, preprocessed, biomarkers, etc.). We recommend users explore this tool for additional analyses beyond enrichment.
For MS1-based metabolomics datasets, particularly in imaging MS, the inherent molecular ambiguity in metabolite identification complicates downstream analyses, including enrichment. To our knowledge, no other metabolite enrichment method addresses isomeric/isobaric ambiguity in enrichment analyses.
In this notebook, we demonstrate how to use S2IsoMEr for
ORA enrichment in single-cell metabolomics datasets while accounting for
isomeric/isobaric ambiguity.
Dataset
The single-cell dataset used is from the SpaceM paper, which models NASH by stimulating Hepa-RG cells with fatty acids and other inhibitors compared to a control, followed by MALDI imaging MS.
The data is freely available in MetaboLights.
Download single-cell matrices and associated metadata
NASH_scm contains the single-cell metabolite matrix
which will be main input as well as condition per cell in
NASH_scm$metadata. These are the main required files to run
single-cell metabolomics enrichment. condition_metadata
contains the METASPACE dataset
names for each replicate, while metaspace_annotations
contains the annotation results for each dataset in the SpaceM
project on METASPACE. We will use the annotation results and
corresponding FDR thresholds to select metabolites as input query and
corresponding universe for enrichment.
NASH_scm_tmp = tempfile()
download.file("https://zenodo.org/records/13318721/files/NASH_scm_dataset.rds", destfile = NASH_scm_tmp)
NASH_scm = readRDS(NASH_scm_tmp)
condition_metadata_tmp = tempfile()
download.file("https://zenodo.org/records/13318721/files/spacem_scm_matrices.rds", destfile = condition_metadata_tmp)
condition_metadata = readRDS(condition_metadata_tmp)[["metaspace_dataset_names"]]
condition_metadata$dataset_name = sub(".ibd.*", "", condition_metadata$dataset_name)
metaspace_annotations_tmp = tempfile()
download.file("https://zenodo.org/records/13318721/files/SpaceM_metaspace_ds_annotations.rds", destfile = metaspace_annotations_tmp)
metaspace_annotations = readRDS(metaspace_annotations_tmp)Prepare the input data
scm = NASH_scm$scm %>%
as.matrix() %>%
t()
conds = NASH_scm$metadata %>%
column_to_rownames("Cell")
conds = conds[colnames(scm),]
conds_unique = conds %>%
dplyr::distinct()
metaspace_annotations = metaspace_annotations %>%
dplyr::left_join(condition_metadata, by = c("ds_name" = "dataset_name")) %>%
dplyr::rename("Replicate" = "Condition") %>%
dplyr::left_join(conds_unique)Filter metabolites and specify conditions
Here we specify the reference and query conditions as
cond_x and cond_y, respectively. And since
METASPACE provides FDR-controlled annotations, we will select
annotations passing desired_fdr as query and all detected
annotations for a given annotation database
(desired_annot_db) as custom universe.
cond_x = "U"
cond_y = "F"
desired_fdr = 0.1
desired_annot_db = "HMDB"
annots_des_fdr = metaspace_annotations %>%
dplyr::filter(Condition %in% c(cond_x, cond_y),
fdr <= desired_fdr,
str_detect(db, desired_annot_db)) %>%
pull(formula_adduct) %>%
intersect(rownames(scm))
custom_univ = metaspace_annotations %>%
dplyr::filter(Condition %in% c(cond_x, cond_y),
str_detect(db, desired_annot_db)) %>%
pull(formula_adduct) %>%
intersect(rownames(scm))
input_scm = scm[annots_des_fdr,]Initialize enrichment object
The first step is creating a S2IsoMEr object which
contains the input matrix in scmatrix and the conditions
for each cell specified in conditions. We need to specify
the enrichment_type as “ORA”. annot_db
corresponds to the annotation database used during the annotation, more
relevant if annotation is performed using METASPACE. The databases supported
are (“CoreMetabolome”, “HMDB”,“SwissLipids”,“LipidMaps”), if you want to
provide a custom annotation database, you can provide it using
annot_custom_db argument. Here we will use “HMDB” as
annotation database.
Since we want to consider isomeric/isobaric ambiguity, we set either
consider_isomers or consider_isobars to
TRUE. If both were set to FALSE, it will run
classic ORA with no bootstrapping. We also specify polarization_mode to
positive since these datasets were acquired in
positive mode.
For enrichment background, we use the background_type
argument to select one of the possible background types :
-
LION: Uses LION ontology. Only for Lipids. - The following types are curated from RAMP-DB 2.0 :
-
super_class: Most coarse-grained classification -
main_class: Fine-grained sub classification compared tosuper_class -
sub_class: Most fine_grained sub classification. -
pathways: Biological pathways curated from KEGG, HMDB, Reactome and WikiPathways.
-
We also specify the background molecule_type by
specifying either Metabo for metabolites or
Lipid for lipids. To pull the relevant background which is
internally built in initEnrichment, you can provide the
previous arguments to the Load_background function to get
list of terms and their associated molecules as follows :
bg = Load_background(mol_type = "Metabo",
bg_type = "sub_class",
feature_type = "name")Finally we specify condition.x and
condition.y as reference and query conditions,
respectively. While running ORA in Run_enrichment, fold
changes will be computed in condition.y relative to
condition.x.
ORA_boot_obj = initEnrichment(scmatrix = input_scm, conditions = conds$Condition,
enrichment_type = "ORA",annot_db = "HMDB",
consider_isomers = T, consider_isobars = T,
polarization_mode = "positive",
background_type = "sub_class",
molecule_type = "Metabo",
condition.x = cond_x,
condition.y = cond_y)There are additional arguments to initEnrichment that
could be used depending on your dataset metadata and any other prior
knowledge. Check documentation of ?initEnrichment for more
information on these arguments.
Running ORA bootstrapping-based enrichment
Based on the initialized object, we provide
Run_enrichment function as a wrapper around the main
enrichment functions :
So you can also provide additional arguments to
Run_enrichment from the argument list of the relevant
function from the above list.
In this example, Run_enrichment will first calculate log
fold change to separate metabolites into upregulated and downregulated
if Run_DE is set to FALSE. If it was set to
TRUE, the function seurat_WilcoxDETest (a
wrapper for WilcoxDETest
from Seurat) will be used to calculate p-values based on a wilcoxon
rank-sum test which will be used in addition to the previously computed
log fold changes to select the markers for ORA based on the
DE_pval_cutoff and DE_LFC_cutoff arguments in
the Run_enrichment function.
The min.pct.diff argument of 0.1 specifies that a marker
must have at least a 10% difference in detection between cells in both
conditions to be considered differentially abundant.
From the additional list of arguments to
Run_bootstrap_ORA we recommend defining the following
arguments to Run_enrichment :
custom_universe: List of background metabolites to be used. More specifically, these represent the set of all possible metabolites that were measured in a given dataset (optionally under a given threshold). If not provided, all metabolites in the selected background will be used and it might lead to potentially misleading results.report_ambiguity_scores: Useful to understand the degree of isomeric/isobaric ambiguity per metabolite.n_bootstraps: Number of bootstrap iterations. The default is 50, but increasing this number generally improves accuracy, though it will slow down the process. Adjust according to your needs. We recommend a minimum of 50 and a maximum of 1000. 100 is acceptable.
ORA_boot_res = Run_enrichment(object = ORA_boot_obj,
custom_universe = custom_univ,
report_ambiguity_scores = T,
DE_LFC_cutoff = 0,min.pct.diff = 0)Output
The main output of Run_bootstrap_ORA is a list of 2
dataframes :
“unfiltered_enrich_res” (
ORA_boot_res[["unfiltered_enrich_res"]]) : Data frame containing the enrichment results for each term and bootstrap and the contingency table used for ORA.“clean_enrich_res” (
ORA_boot_res[["clean_enrich_res"]]): Summary statistics per term passing the filters specified inRun_bootstrap_ORA
Check ?Run_bootstrap_ORA for more information on the
output and other parameters.
Since we used Run_enrichment as a wrapper around
Run_bootstrap_ORA we get the ORA results separately for
upregulated, downregulated, and
all metabolites based on the calculated
DE_LFC_cutoff specified above. If you use
Run_bootstrap_ORA directly, you can provide a list of
multiple conditions as input directly as long as you know the markers
a priori.
To get a summary of the terms passing each filter, we can call
passed_filters_per_term function on any unfiltered
dataframe and any combinations of filters to understand why a given term
was excluded in the final summarized results. Check
?passed_filters_per_term for more information on the
filters.
enrich_ORA_summary = passed_filters_per_term(unfiltered_df = ORA_boot_res$upregulated$unfiltered_enrich_res,
enrich_type = "ORA", min_intersection = 3,alpha_cutoff = 0.05,q.val_cutoff = 0.2,boot_fract_cutoff = 0.5)
enrich_ORA_summary = enrich_ORA_summary[order(enrich_ORA_summary$pass_all_filts, decreasing = T),]
head(enrich_ORA_summary)Visualization
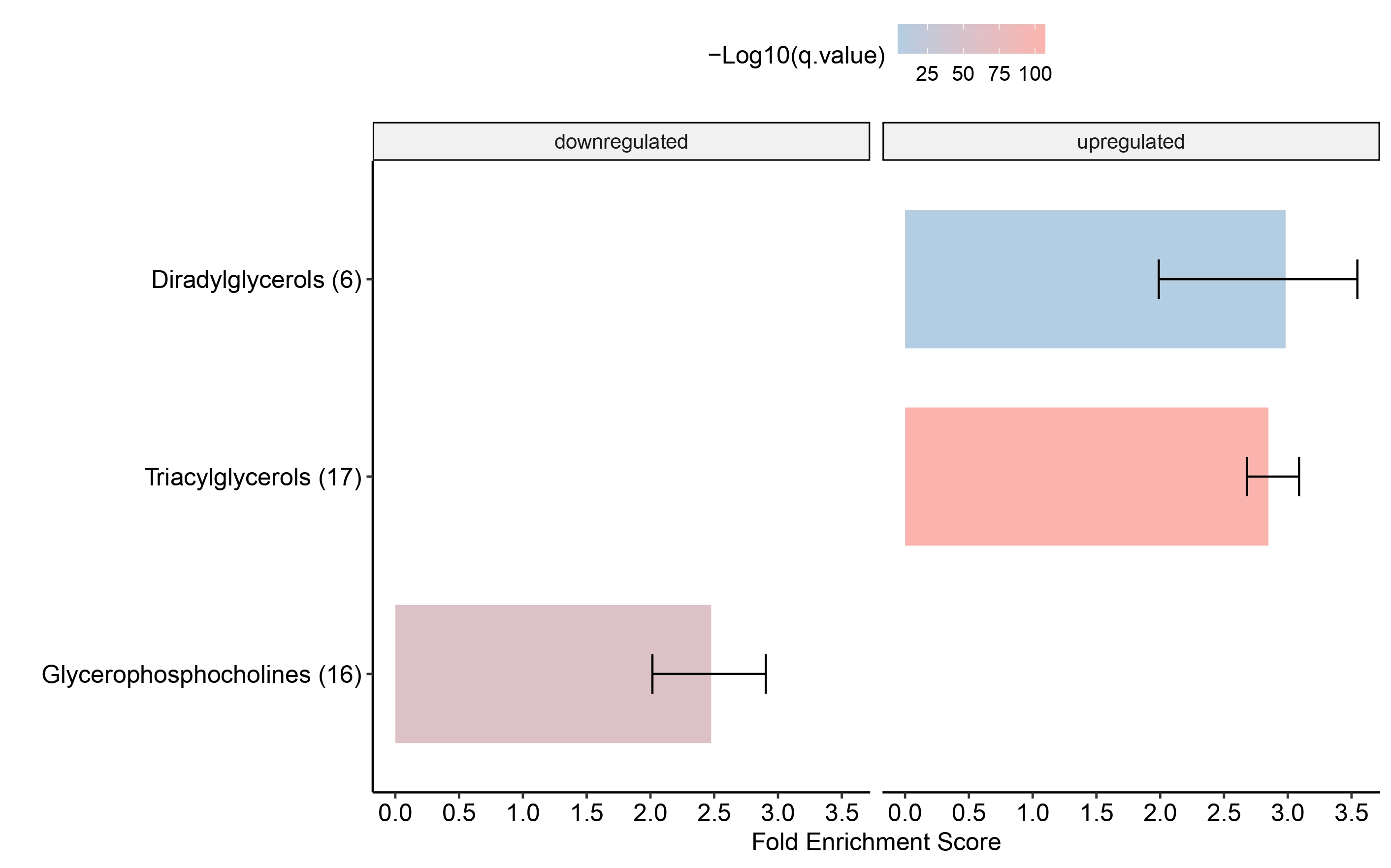
Dotplot
multi_cond_collapse = collapse_ORA_boot_multi_cond(ORA_boot_res_list = ORA_boot_res)
dotplot_ORA(ORA_res = multi_cond_collapse$clean_enrich_res)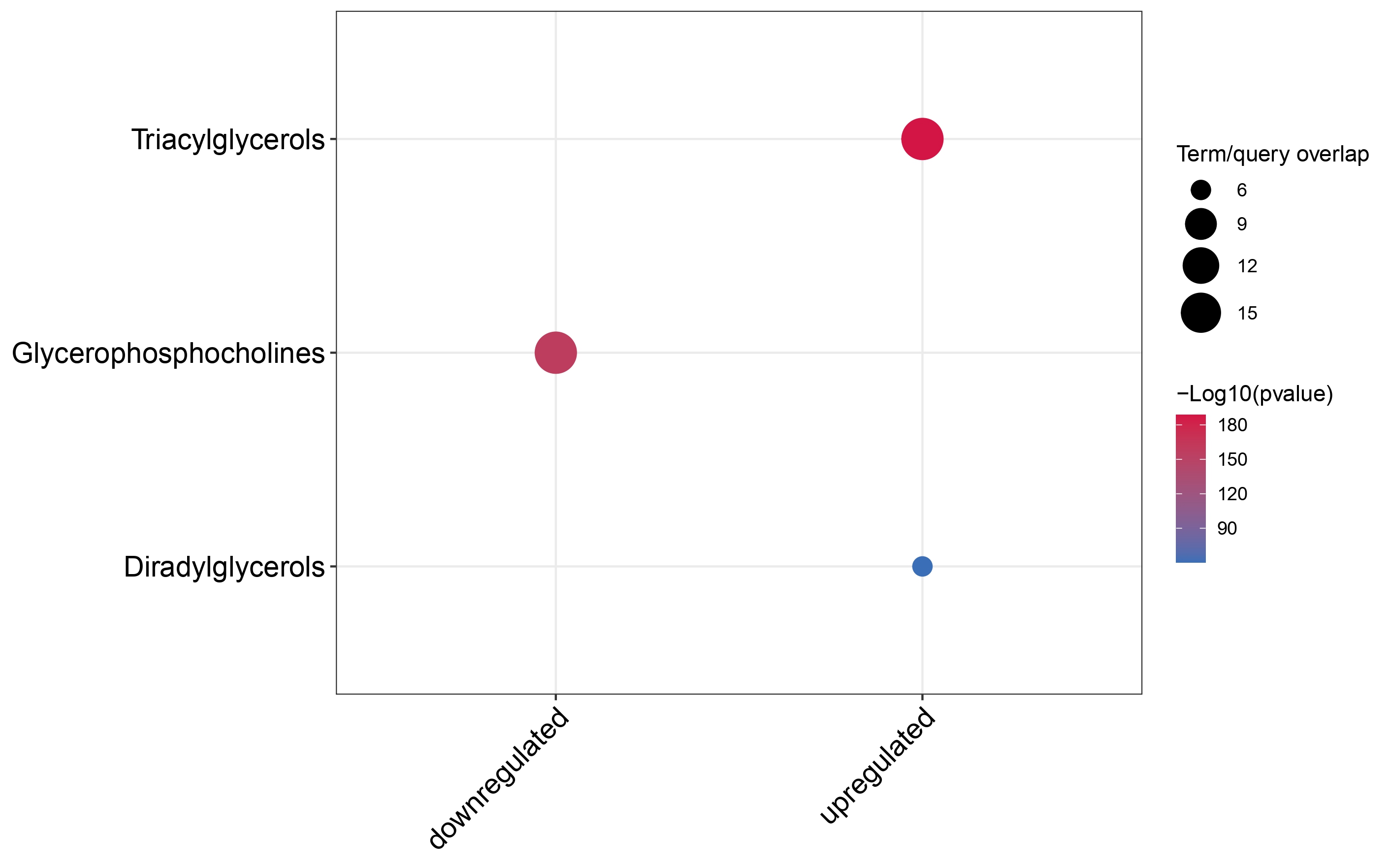
Ridge plots
To compare the distribution of term/query overlap size across
bootstraps we can plot the distribution of the overlap size across terms
of interest. Here we use ridge_bootstraps function to plot
the distribution of the terms enriched in both upregulated and
downregulated markers found in multi_cond_collapse using
the upregulated results only.
ridge_bootstraps(enrich_res = multi_cond_collapse$unfiltered_enrich_res,
terms_of_interest = c(multi_cond_collapse$clean_enrich_res$Term),
condition = "upregulated")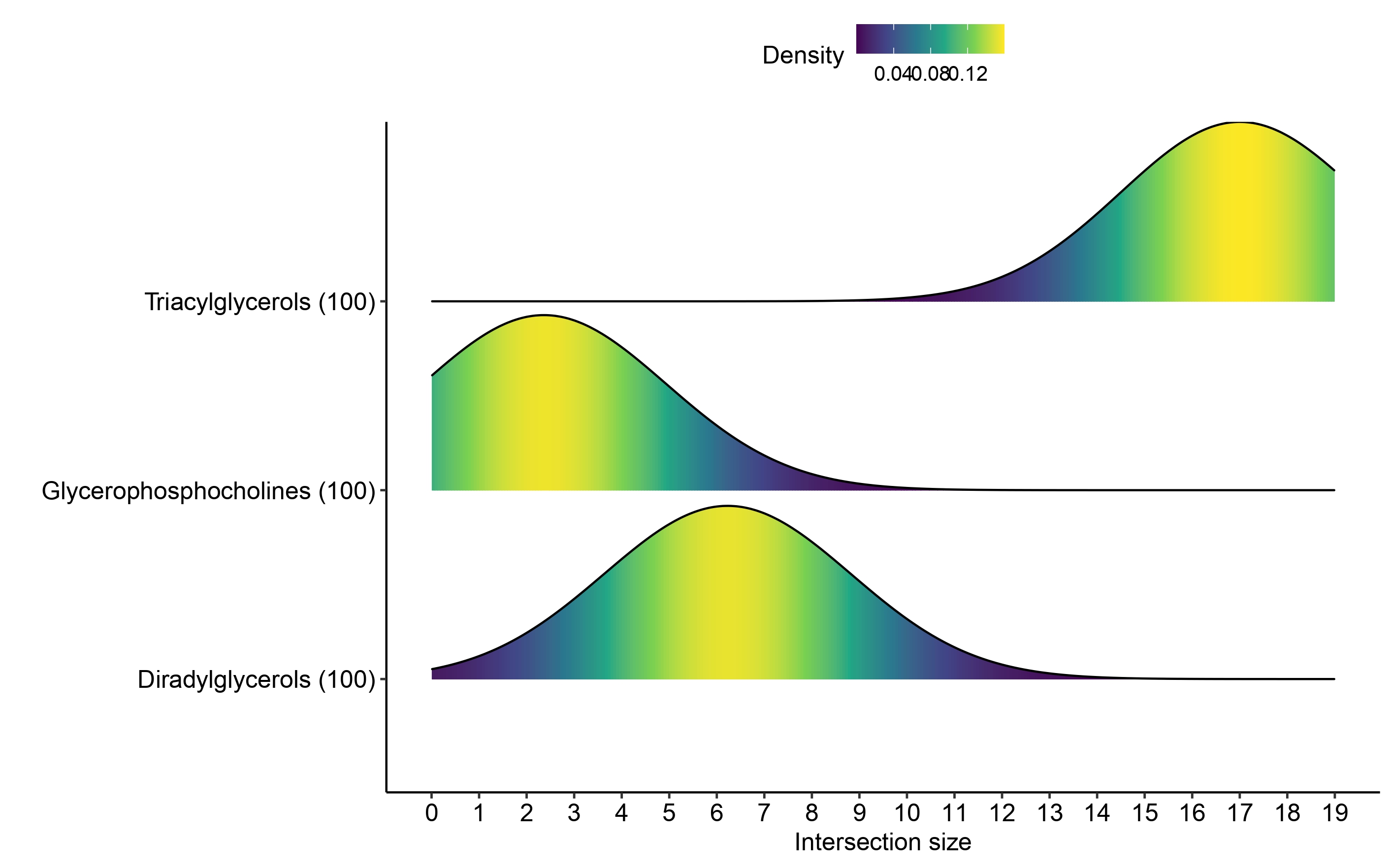
Comparative distribution of marker ions
We also provide a simple function to get the TP markers associated
with a given term which correspond to the input ion in the
input_scm matrix used as input.
TP_ions = get_TP_markers_per_Term(ORA_boot_df = ORA_boot_res$downregulated$unfiltered_enrich_res,
term_of_interest = "Glycerophosphocholines")
TP_ions = map_TP_markers_to_ions(markers = TP_ions,
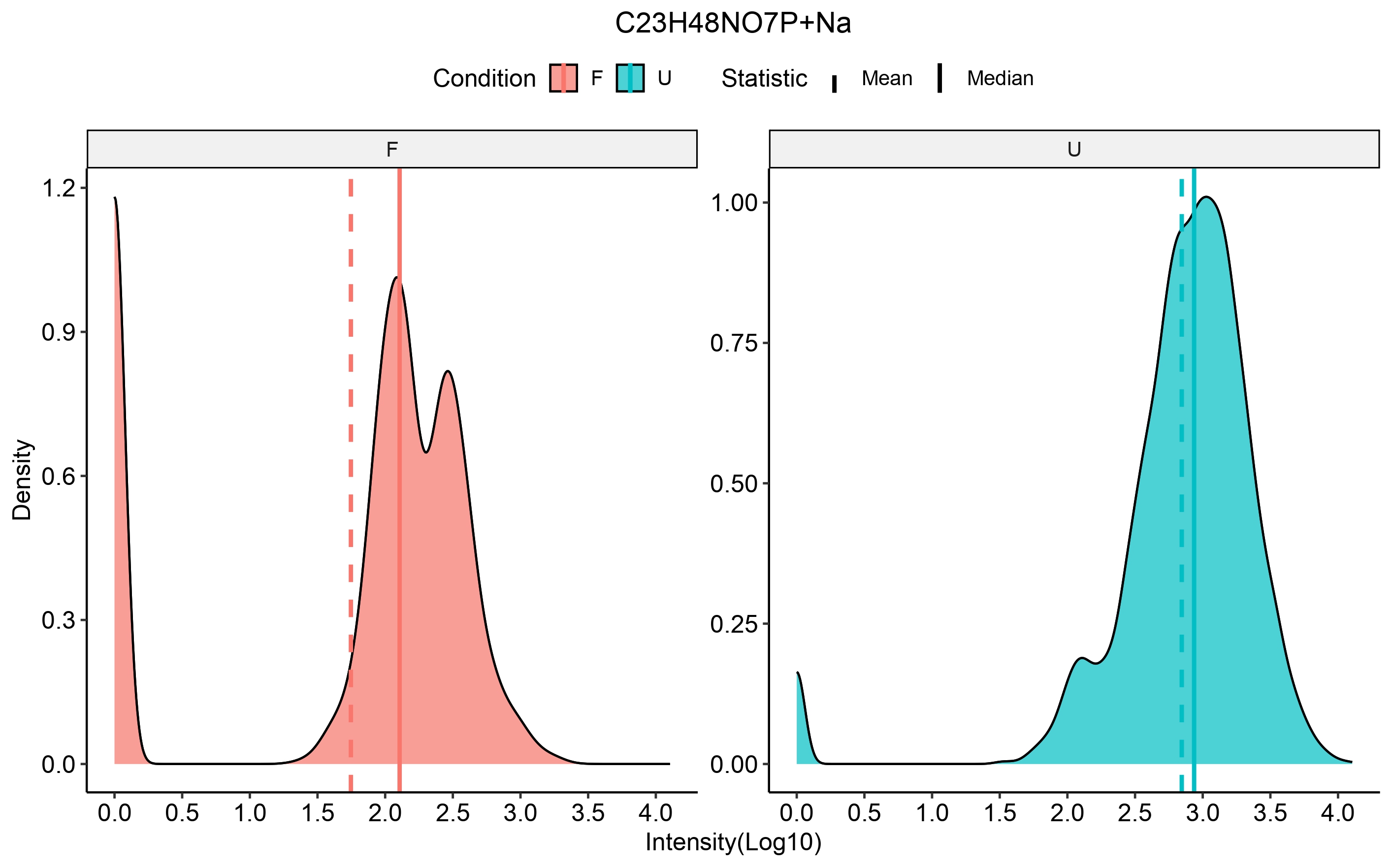
scm_ions = rownames(input_scm))We can then select an ion of interest to check distribution of
intensities in the specified conditions using the
compare_metabo_distr function which takes the
ORA_boot_obj as input, the ions and the conditions of
interest.
compare_metabo_distr(ORA_boot_obj, metabolite = TP_ions[5],
conds_of_interest = c(cond_x, cond_y))